Drug-Likeness and RNA-Targeted Small Molecules
Thomas Hermann, Associate Professor of Chemistry and Biochemistry at University of California, San Diego and Co-Director, UCSD Center for Drug Discovery Innovation

Only a minuscule fraction of the human genome is devoted to encoding proteins, yet proteins provide nearly 100% of the targets for currently marketed drugs. The few exceptions include antisense and RNAi oligonucleotides, some DNA-binding cancer therapeutics such as doxorubicin and cisplatin, and a chemically diverse group of antibiotics that interfere with bacterial protein biosynthesis by targeting selectively the RNA components of the pathogens’ ribosomes.
This latter example, of antibiotics acting on bacterial RNA, inspired a hunt for new drugs targeting viral RNA to combat the HIV epidemic in the last decade of the 20th century. The viral genome contains several noncoding RNA (ncRNA) motifs that are essential for the infection process. High conservation in HIV clinical isolates of ncRNA sequences, such as the trans-activation response (TAR) and Rev response (RRE) elements, offered targets with a high barrier to mutational resistance and spurred drug discovery efforts both in pharma industry and academia. Eventually, the notion of RNA as a drug target congealed around these two HIV RNAs, inspiring researchers to broaden their horizons in exploring non-traditional targets among ncRNA elements.
The lure of the as-yet-unexplored world of RNA targets, beyond the familiar confines of proteins, made it tempting to surmise that a new class of RNA-friendly molecules might be required to tackle this new frontier: Exceptional targets call for exceptional ligands. Accordingly, for the better part of the last two decades, the design of RNA-binding ligands focused on molecules that combined motifs known to confer affinity to nucleic acids via intercalation, groove binding, and contacts with charged phosphate groups. But these motifs typically proved to be incompatible with guiding principles of drug-likeness. In the heat of the battle to conquer RNA as a drug target, ligand designers committed the cardinal sin of medicinal chemistry (the author, too, guilty as charged). This design strategy was predicated on the hope that one would be able to design away from poor drug properties later if only a potent ligand was found initially, drug-likeness be damned. A notion took hold declaring it acceptable to forgo established concepts of drug-likeness for the sake of making inroads with a difficult new target class. After all, a majority of RNA-binding antibiotics are natural products or derivatives thereof which break common rules of drug-likeness, from limits on molecular mass, the number of hydrogen bond donors and acceptors, to reasonable lipophilicity.
What was often downplayed in this strategy, however, is that most of the known RNA-binding antibiotics occupy singular niches. Some RNA-binding antibiotics never make it into eukaryotic cells, relying on active transport mechanisms that take them into bacterial cells. A number of them are administered only as injectables. Some RNA-targeting antibiotics are textbook examples of drug-likeness: Linezolid, an RNA-binding oxazolidinone inhibitor of bacterial protein synthesis, has an oral bioavailability of close to 100%, a molecular mass of under 350, and a biological half-life of 5h. A more drug-like molecule is hardly imaginable. It is molecules like linezolid that will be dependable lodestars of drug discovery for RNA targets.
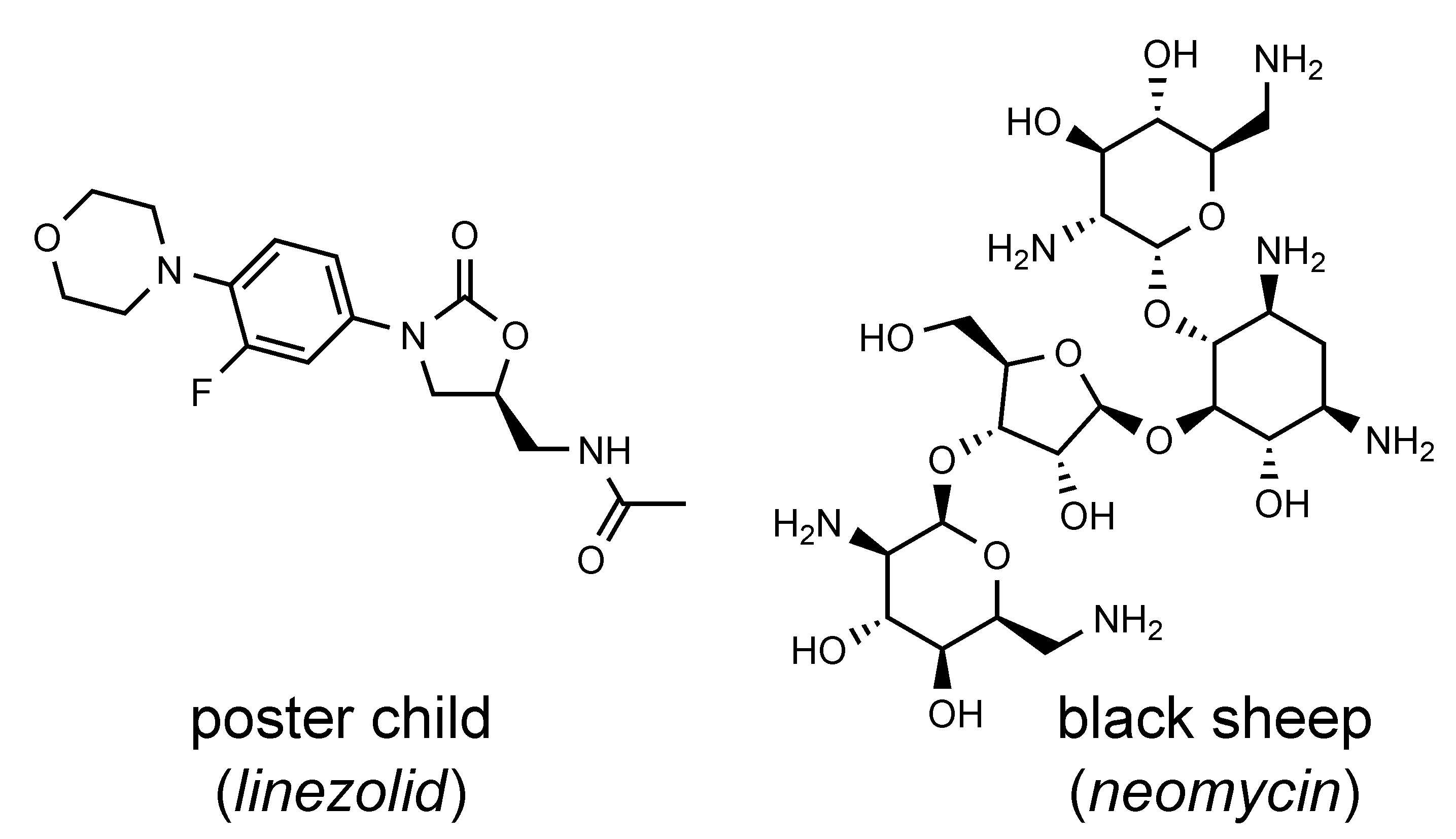
This should be reassuring to both screeners and medicinal chemists. There is no molecular pixie dust to be invoked in the search for RNA-targeted small molecules (rSMs). Certainly, screening libraries should have room for molecules that exploit idiosyncrasies of known RNA binders, including functional groups that allow for interactions with metal ion and hydration sites or intercalation between bases. Yet, for selecting molecules for screening, chemical diversity and drug-likeness ultimately are still king. From the perspective of the RNA targets, familiar concepts of medicinal chemistry can be exploited, such as the optimization of ligand shape complementarity and utilization of hydrophobic pockets. Counterintuitive as this may seem for such a hydrophilic macromolecule, some RNA architectures may fold to form non-polar patches and dry pockets which stand out all the more as selective recognition sites among a sea of polarity and charge.
The exceptional promise of RNA as a drug target derives from the fact that no other class of biological macromolecules can lay claim to represent potentially 90% of the human genome. This enormous opportunity deserves to be met with an adventurous disposition, employing novel approaches of target discovery, validation, screening and mechanistic studies. Anything goes in the lab. Yet, when it comes to discovering and developing RNA-targeted drugs, the operative focus ought to be on keeping the ligands drug-like, no matter how tempting it is to give in to the short-term problem of simply binding to a compelling target.