Arrakis achieves escape velocity
Michael Gilman, PhD, CEO, Arrakis


When last we spoke (here and here), Arrakis had just achieved liftoff with a $38M Series A financing in February 2017, and we were awash in fascinating and, in some cases, vexing questions. Two of my own questions that stand out upon rereading those posts from two years ago are:
Is medicinal chemistry truly limited to a small slice of the proteome, or are we being held back by our own orthodoxy?
Can we reimagine small molecule drug discovery, re-architect the drug discovery toolkit, and point it at RNA instead of proteins?
Those questions, for me at least, were deeply fascinating. Among the vexing questions were:
RNA is floppy. Does it even have structure, let alone druggable pockets?
RNA only has four bases. How can you ever expect to achieve molecular selectivity?
RNA inside a cell is clothed in RNA-binding proteins. Does the structure of a naked RNA in a test tube even remotely resemble its state in a living cell?
And many more, which all more or less integrated into:
Are you guys nuts?
Well, we’re not (or if we are, it’s for completely unrelated and largely nonprofessional reasons). In fact, as we take Arrakis to escape velocity, with a $75M Series B financing announced today, we know that the answer to all of these questions (except for the one questioning our sanity) is resoundingly yes. And we’ve set a new course: Destination patients!
A bit of prehistory
Before I explain, let’s revisit why we embarked on this mission.
Pretty much our entire pharmacopeia, the collection of medicines we take to treat disease, is aimed at proteins. The conventional argument for why proteins is that they are the picks and shovels of the cell – the molecules that do the real work. My own hypothesis is that our industry’s overarching focus on proteins is largely a historical artefact of the roots of the modern pharma industry in a time when proteins were thought to be the only important molecules in the cell. Indeed, prior to the Hershey-Chase experiment in 1952, most scientists believed that genes must be encoded by proteins, because nucleic acids were too boring and information-poor to do the job.
Now, of course, we know better. We know that DNA is where genes live and that RNA executes those instructions. As Eminent Founder Jennifer Petter constantly reminds us:
RNA is upstream of all biology!
If we knew then what we know now, maybe we’d have skipped proteins entirely – they’re challenging to purify, nearly impossible to synthesize, and opaque inside cells – and gone right after RNA.
But it’s not too late. Our vision at Arrakis is to unlock all of the biology of the cell for medicines by rebuilding the industry’s powerful small-molecule drug discovery toolkit to point it at RNA. Our goal is to develop a new class of medicines, RNA-targeted small molecules – rSMs – orally-bioavailable drugs that impact the biology of specific RNAs.
Today, as we embark on this expanded mission, I am providing you with an interim report.
The yin-yang of structure and function
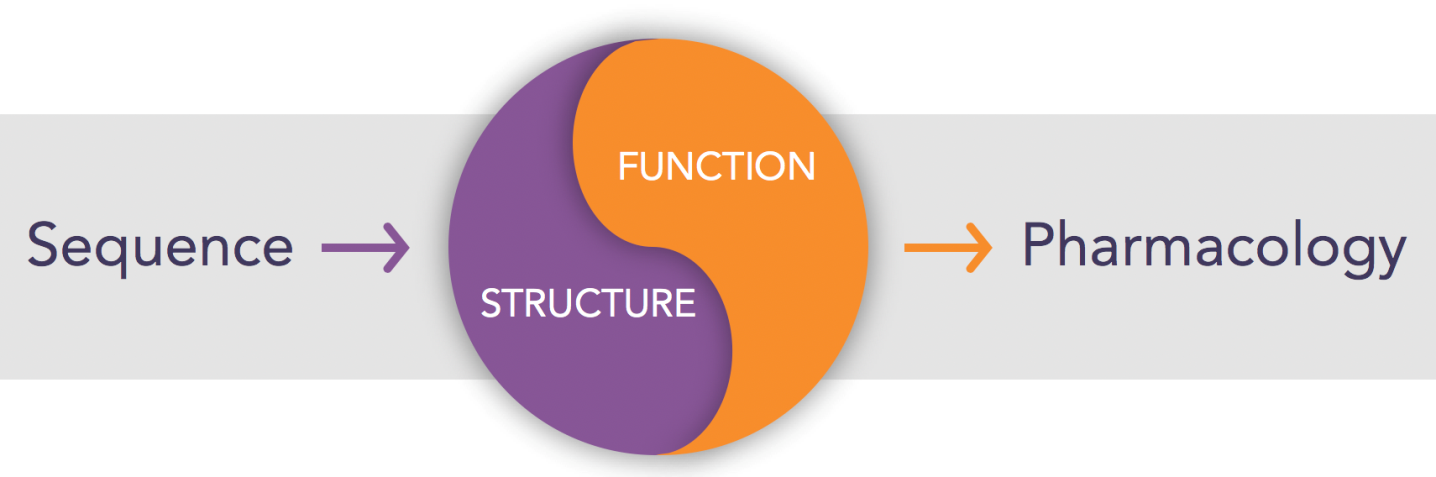
Our success as an industry at drugging proteins is based on a deep understanding – so deep that we take it for granted – of the relationship between primary sequence and three-dimensional structure and between structure and function. We can look at a protein sequence and instantly identify it as a protein kinase, for example. Once we know it’s a kinase, we know largely how it folds, where the active site sits, what it does, and how to assay its activity. And we can quickly get to work at finding an inhibitor.
For RNA, we simply did not have this intellectual framework. Can we infer two- and three-dimensional RNA structure from primary sequence? Are there consensus structures or common motifs, as we generally have for proteins? What functions are encoded in RNA and are they associated with specific, recognizable structures? One thing that’s clear about RNAs: They don’t have active sites like enzymes do. Instead, it’s likely that active rSMs will function like allosteric inhibitors, interfering with an RNA’s ability to attain a state required to do its job. The whole concept of drugging RNA is orthogonal to how we think about drugging proteins.
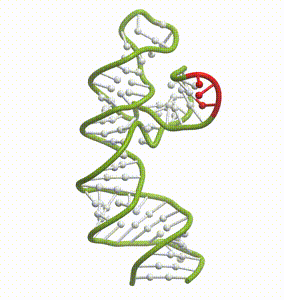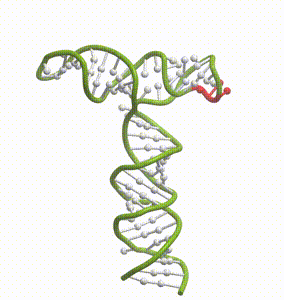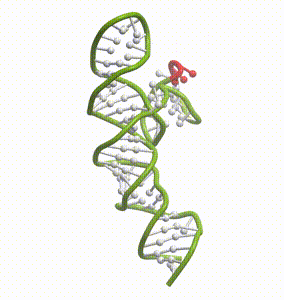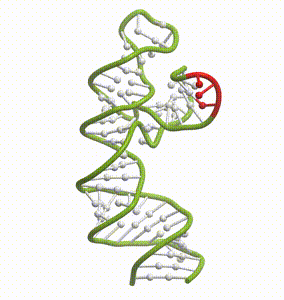
DeepFolding solutions–Arrakis’s proprietary method for 3D RNA folding–for an mRNA structure spanning the AUG start codon (red)
So we had to get our heads around all of that. And then we had to get to work building the tools to answer these questions. We built and refined algorithms to predict two-dimensional folds and three-dimensional structures. We developed methods to recognize pockets with drug-friendly biophysical properties. Importantly, we gave a lot of weight to understanding the dynamics of RNA targets. Is there one predominant stable structure? Several related structures in rapid equilibrium (left)? Hopelessly floppy strands of spaghetti? Can we recognize “domains” in these RNAs that have an optimal degree of dynamics for ligandability?
Then we had to go into the lab and make sure the computers were telling us the truth. We have substantially expanded Kevin Weeks’s SHAPE approach to validate folding predictions and, importantly, have now generated data on the structure of fully folded, mature protein-bound mRNAs for our top fifty target genes in living cells. We know what structures are faithfully recapitulated in the test tube and which ones may vary, perhaps owing to differences in dynamics inside the cell.
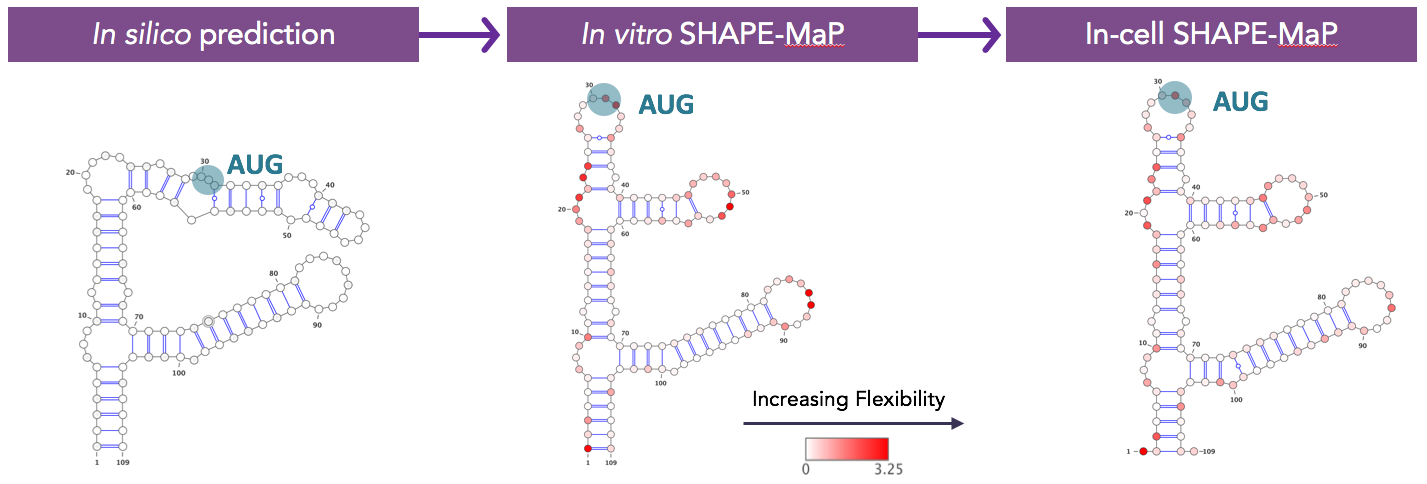
Predicted two-dimensional structure of the same mRNA purely in silico, constrained by SHAPE-MaP data on in vitro folded RNA fragment, and constrained by SHAPE-MaP data on the mature mRNA in living cells
To assign function to these structures, we’ve exploited many rich public databases to identify regions of target RNAs that seem to be loci of biology. We are mapping where RNA-binding proteins interact with key targets and where ribosomes struggle to pass. And we’ve developed efficient high-throughput laboratory methods for validating the contribution of specific structures and interactions to RNA function.
All of this, over the last two years, has enabled us to pivot from a largely empirical approach to rSM discovery to a strategy that is satisfyingly hypothesis-driven.
The awesome power of RNA synthesis and sequencing
A signal advantage of working with RNA rather than proteins is that RNA is easy to synthesize. In theory.
In practice, though, producing RNA in sufficient quantity and quality to support structural studies, screening, and assay development is not for the faint-hearted. And to do so day after day after day requires both a deft touch and an attention to detail that would elude most mortals. But our exceptional RNA team has mastered the art and science of RNA synthesis at scale, using an impressive combination of creativity, meticulousness, and old-fashioned elbow grease. Moreover, they’ve taught RNA to perform amazing new tricks by incorporating proprietary chemical biology reagents into RNA molecules, enabling us to install new functionalities into RNA to facilitate screening and assay development.
Another delightful feature of RNA is how easy it is to sequence. Even better, the supra-Moore’s law reduction in the cost of next-generation sequencing (NGS) enables us to use sequencing as a routine assay read-out. We’ve generated roughly 20 terabytes of sequencing data on our mission to date, the lion’s share in our massive determination of in-cell mRNA folds for 50 target genes across 30 cell lines.
The combination of chemical biology and NGS will really pay off as our programs progress, as our PEARL-seq platform will allow us to interrogate target engagement and in-cell selectivity of our drug candidates with depth and fastidiousness that will make old-school protein hands swoon with jealousy.
Mining for spice
Once we’ve identified a structure of interest, where do the compounds come from? Here we’ve tried, wherever possible, to repurpose established screening tools. But, of course, making these methods RNA-friendly can be quite a job. It’s not just resurfacing your kitchen cabinets and buying a shiny new internet-connected refrigerator. In the case of DNA-encoded libraries
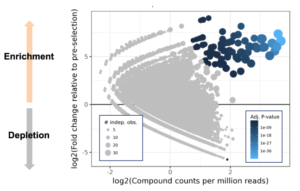
Sample output from DEL screen. Blue dots represent putative hits
(DELs), we worked with our collaborators at Vipergen to rip their system right down to the studs. We changed everything – incubation conditions, time, temperature, volume, the DNA tags, the statistical analysis, even the libraries. We ran close to six hundred screens to get it right and at peak were screening fifteen RNAs a week. Because the marginal cost for putting a target into the hyper-efficient machine we’d built with Vipergen was so low, we could afford to use it empirically – for pocket discovery. If a target returned hits, we knew it had a pocket.
But as we developed more conviction about targets – where the druggable structures and functions lie in high-value RNAs – we could afford to turn to methods where the throughput is lower but the quality of the chemistry is higher. The chemistry of DELs is, by definition and necessity, challenging. At Arrakis we are ruthlessly committed to building on the decades of learnings on what makes a small molecule compound an excellent pharmaceutical. No need to make things harder for ourselves. Consequently, we want to begin our quest with compounds that are as drug-like as possible. That’s our focus today – screening libraries of highly drug-like compounds through a technically complex but biophysically straightforward method. And we’ve had great success so far. We’re getting hits to every target we screen. These hits are remarkably bland to look at – which is a good thing; they are delightfully druglike (below left). They have affinities typical of protein screens, low µM or better, and, importantly, are selective for the RNA we’ve screened (below right). Along the way, we’re learning the dos and don’ts of RNA screens and are using those lessons to build a highly curated internal compound library, rich in RNA-friendly scaffolds and rigorously purged of troublemakers, of which there are plenty in most libraries.
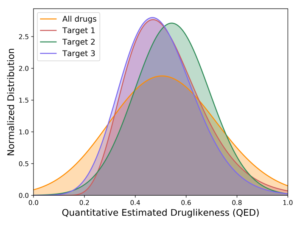
Estimation of druglikeness of primary hits from three screens relative to known drugs
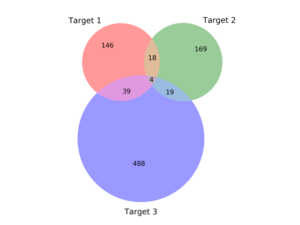
Relative selectivity of primary hits against three RNA targets
We’ve now launched several medicinal chemistry programs against targets that I’ve got to say are pretty fabulous. Our most mature hit-to-lead program is four or five months old. I think I’d summarize our experience here by saying that SAR for RNA is about 80% like SAR for proteins and 20% breathtakingly bizarre.
Destination patients
So now, thanks to the remarkable and dedicated effort of the intrepid team at Arrakis and the support of our visionary investors, we are poised to realize the promise of bringing RNA into play for small-molecule drug discovery. We have a burgeoning list of targets that we are truly excited about. Many of these targets are in cancer, where there is a rich stew of opportunity, ranging from oncogenes and transcription factors, whose role in cancer has been known for decades, to hipster targets emerging from genetic and genomic screens for driver and synthetic lethal genes. We’re also interested in targets in other therapeutic areas that are highly validated genetically or clinically but basically undruggable any other way.
Over the past two years, Arrakis has built an end-to-end platform for discovering rSMs and we can now begin to make good on our vision of a world in which all RNA biology can be accessed to bring new medicines to millions of patients.