Why NOW for Targeting RNA with Small Molecules?
Jennifer Petter, PhD, Founder and Chief Scientific Officer
In “The Right Stuff,” Tom Wolfe noted that astronauts were granted hero status before their missions, perhaps because no one expected them to return. Happily, Arrakis also has received a lot of laudatory attention just for launching the mission of drugging RNAs with small molecules – before we’ve even achieved it. The plaudits are lovely, and we’re pretty sure we’ll survive that ordeal, but there is still the question: Why is this mission considered so challenging? Is it because others, who were just as bold and inquisitive as we are, have tried and failed? I doubt it.
Sure, we’ve got experienced people and a solid plan. But we believe a lot of our success will also derive from great timing. The world has changed in some important ways, making our path forward more clear.
A Tougher Task?
Before tackling the question of why everyone thinks drugging RNA is so hard, one might ask why drugging proteins is considered to be so easy (at least by comparison). Put differently, why were proteins first?
Well, the study of protein structure and function actually significantly antedates that of RNA. Many of the readily studied gross observations in biochemistry and pharmacology can be directly traced to protein action – including human genetic diseases derived from mutations that impact protein structure and function. Moreover, biochemistry quickly uncovered the mechanisms of action of endogenous small molecules (co-factors, substrates, metabolites, etc.). And then came x-ray crystallography of proteins, and the ability of molecular biology to MAKE proteins.
But during all this excitement about proteins and interdicting their function for therapeutic benefit, RNA has been relegated to a comparatively passive role: DNA makes RNA makes protein. The whole POINT of the central dogma was to make proteins, so what’s the problem?
The problem is that RNA function is complex and often on the critical path to biological interventions we seek.
Early Forays into Therapeutic Targeting of RNA
RNA has already been drugged. Ribosomal RNA was drugged serendipitously with natural product antibiotics and later linezolid. A design-driven approach to drugging RNA was achieved with antisense oligonucleotides (ASOs) and then by RNA interference (RNAi) mechanisms. The original promise of ASOs was that complementary hybridization was based on a modular pharmacophore in the ASO, which in turn could be readily assembled via modular synthesis. So knowing the sequence of interest, one could, in principle, punch the desired sequence into a nucleic acid synthesizer and voila, out popped a drug. Of course, as we all know, it really isn’t quite that straightforward. Leaving aside the many challenges of identifying and optimizing ASOs and RNAi, even after years of valiant efforts to improve these molecules’ pharmaceutical properties, they still suffer from very discouraging shortcomings in manufacturing, administration, distribution, and cellular penetration.
But RNA offers other, less modular pharmacophores – pockets similar to those that are so familiar in proteins. When many of us read our biology textbooks, RNA came in three basic flavors: rRNA (blobs), mRNA (strings), and tRNA (adapters). The stars of the show were clearly the proteins this apparatus makes and very little attention was paid to the RNA itself by the drug design community. In fact, in the dark ages, reading Watson by candlelight, one might get the impression that the RNA in ribosomes was probably just a scaffold to arrange the proteins responsible for translation.
New Dimensions of RNA Unfold
Of course, with the remarkable work of Steitz and others, we now know an enormous amount about not just the structure but also central functional role of RNAs in the ribosome. And over the decades we have discovered more flavors of RNA than there are coffees at Starbucks.
And, most relevant to Arrakis’s mission, it has also become clear that those RNAs fold, a lot. And that that folding is related to the complex functions both carried out by RNAs and the regulatory functions visited upon RNAs. In fact, RNA folding (including mRNA!) is ubiquitous, persistent, dynamic, and conserved. And the specific folded shapes assumed by RNA are often dictated by primary sequence, not imposed by associated RNA-binding proteins (RBPs).
We and others have demonstrated that pockets exist and persist in folded RNAs. Moreover, those pockets appear to be compatible with drug-like small molecules. In fact, a wide range of molecules, many of them natural products, have been discovered that bind to rRNA. Many are natural products – polyamines that are non-specific, toxic, and difficult to administer. Some semi-synthetic and synthetic antibiotics have been identified that bind to rRNA (clindamycin, linezolid). Even more exciting are the very drug-like molecules reported by Merck and Novartis that bind to the FMN riboswitch and to SMN2 pre-mRNA, respectively. But none of these successes arise from a directed and intentional approach to drug RNA structures.
So why are RNA structures considered problematic as drug targets? Let me count the ways:
- People haven’t looked for the pockets and frankly didn’t expect to find them.
- Folded RNA, as mentioned, is dynamic, so there is an expectation that any pocket you find may be poorly structured or ephemeral.
- To the extent that a pocket is largely a groove (minor or major), there is the expectation that binders will not be specific.
- The concern that RNA is much more polar than proteins, so relatively nonpolar ligands won’t bind and that polycationic alternatives won’t penetrate membranes.
- That selectivity will be a stubborn problem because with only four bases rather than 20 amino acids, structural diversity will be low, and there are so many RNAs.
- And the most fundamental question – OK, suppose you do manage to find a pocket and bind to it, so what? Does it impact the function of the RNA in any way? And in a therapeutically interesting way?
Solutions/Advantages
Well, we at Arrakis believe that each of these problems is either NOT a problem (sometimes even an advantage) or that, where the problem is real, solutions are readily available. And, moreover, we believe many of those solutions were NOT available 20, or even as few as 10 years ago. Here are some of them:
- Multiple clinical proof-of-concept successes with oligos. Whatever reservations we might have about their pharmaceutical properties, they have proven effective in many diseases.
- Much-improved appreciation of the ubiquity and functional capabilities of RNA structures. The advent of SHAPE techniques has opened all our eyes to this.
- Invention of in vitro This enables the facile production of many RNAs, making RNA production in many ways more straightforward than protein production.
- Some recent stunning successes from the Merck and Novartis groups in identifying drug-like small molecules that target RNA (which we term “rSMs”).
- Next-generation sequencing – the ability to sequence genetic material at a truly blazing pace for very reasonable prices.
Of the above recent developments, next-generation sequencing (NGS) is the most profound, as it suffuses and enables all the other four. Let’s face it, 20+ years ago, when earlier pioneers targeting RNA/SM interactions started up (Scriptgen, Rib-X, Ribotarget, Biorelix), we were sequencing with stone tools. In targeting RNA, sequencing plays an important role at every step of the drug discovery value chain: target identification, target validation, screening, assay development, selectivity assessment, biomarker development, RNA production and QC, target engagement, molecular mechanism of action.
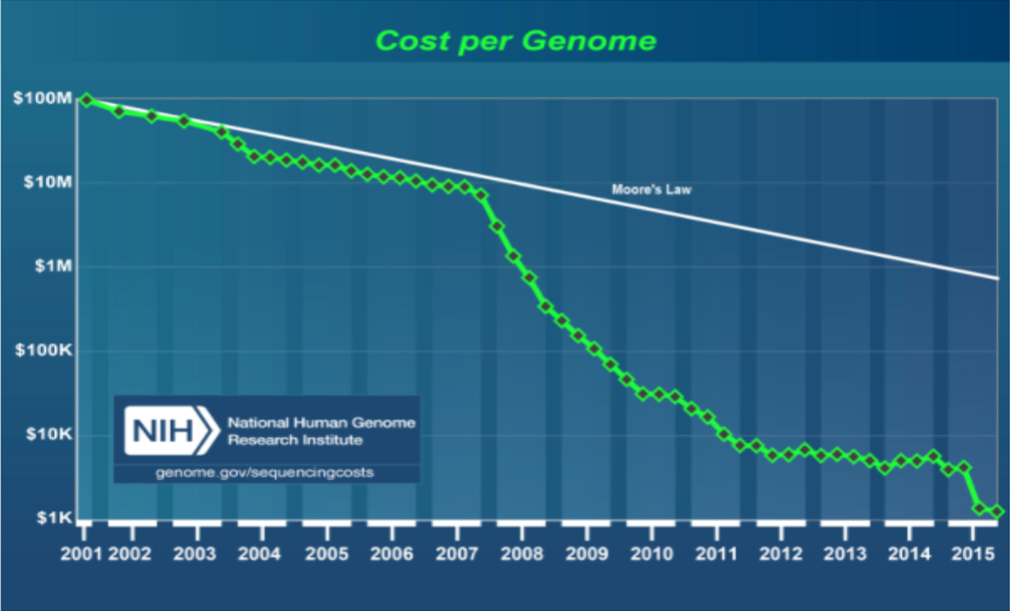
In measuring sequencing power, the consensus metric is what it costs (and how long it takes) to sequence one human genome. Though a 10-fold cost reduction was achieved in the 6 years following from the first complete human genome sequence in 2001, sequencing one genome would cost about $10M, a significant chunk of even today’s hefty Series A rounds. In the ten years since the introduction of NGS in 2007, the cost per genome has dropped another four orders of magnitude. Now, sequencing is so fast and so affordable, at about $1K/genome), an ambitious drug discovery program could use it as a primary assay in a screen (https://www.genome.gov/27565109/the-cost-of-sequencing-a-human-genome/).
At Arrakis, this newfound “superpower” underlies everything we do: Inferring RNA structure from sequence (TRYST), assessing RNA structural parameters in cells, producing large numbers of RNAs to support a broad screening platform, building a chemical biology platform that provides insight into target engagement, binding site, inter-RNA selectivity, and molecular mechanisms of action of a ligand (PEARL-seq). So much of what we need to do to drug RNAs would be impossible without NGS, or at least impossible within the patience horizon of a biotech investor. We often have employed the neologism “re-architecting” to describe how, at Arrakis, we are adapting so many of the tools and methods and concepts of protein/SM drug discovery to RNA/SM drug discovery. The foundation of that new architecture is the stunning speed and affordability of NGS.
Our ambition at Arrakis is to conquer the druggable transcriptome and in so doing find new, breakthrough medicines. This is only conceivable because of the seminal contributions of investigators – both scientific and technical – who have worked to understand in great detail the structure and function and therapeutic potential of RNA.